Testes Clínicos

Programas elaborados para inspirar confiança
Nossos pacotes de dados oferecem evidência adicional dos nossos pontos fortes. A Amgen gera dados robustos e rigorosos sobre cada um dos seus medicamentos biossimilares. Esses pacotes de dados:1
- Usam desfechos e populações de pacientes sensíveis
- Garantem a ausência de diferenças clinicamente significativas em termos de:
- Eficácia
- Segurança
- Imunogenicidade
Esses dados são necessários para aprovação e, o que é importante, equipam os médicos e pacientes com informações de que precisam para tomar decisões de tratamento com confiança.

Dados analíticos, clínicos e não clínicos
Depois de avaliar o medicamento de referência e estabelecer seus atributos de qualidade críticos (CQAs), elaboramos um processo de produção para o biossimilar. Depois, após vários anos de testes, coletamos dados para provar a similaridade e obter aprovação.
- Uma comparação abrangente dos atributos da molécula biossimilar com o medicamento de referência usando tecnologia de ponta garante a ausência de diferenças clinicamente significativas.1,2
- Mesmo com dados analíticos robustos, o biossimilar somente pode ser aprovado com dados clínicos e não clínicos adicionais.2
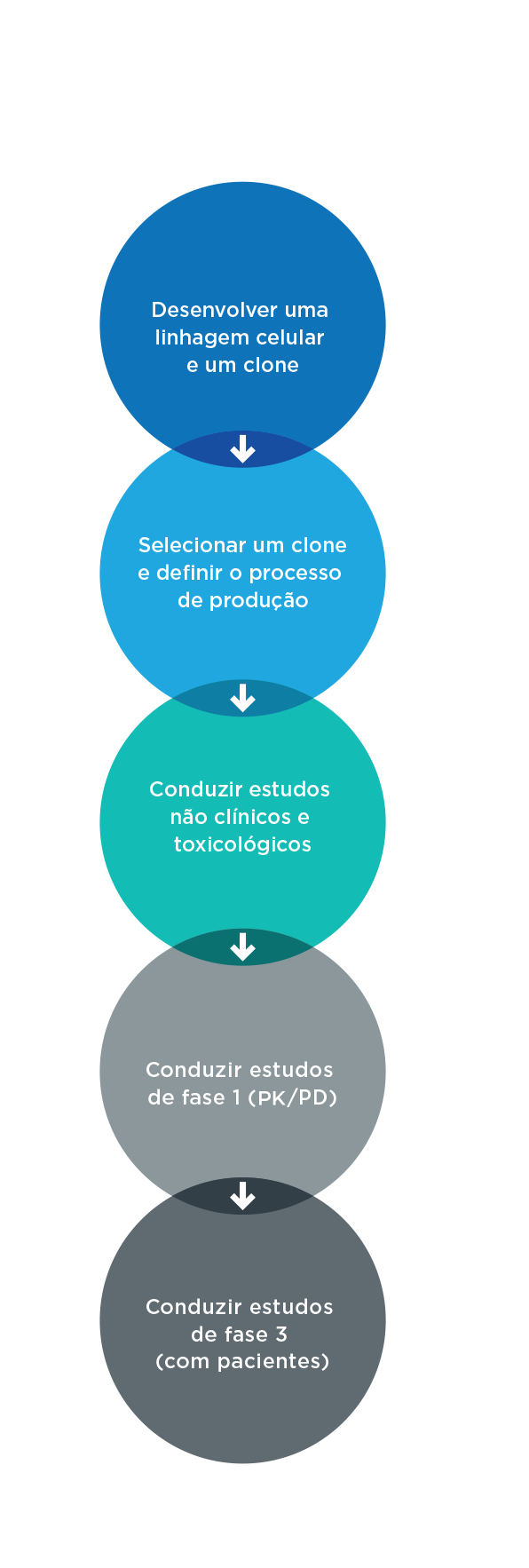
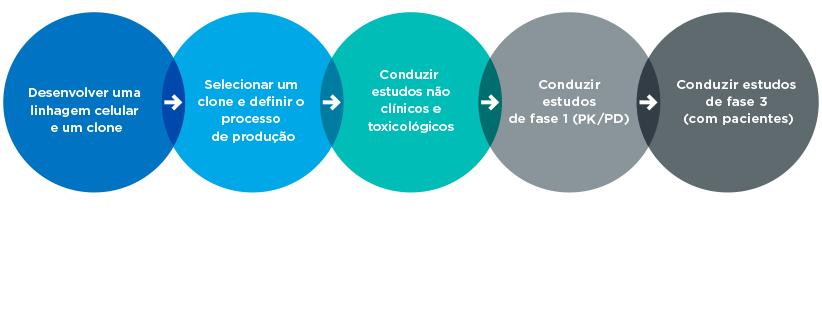
Depois de avaliar o medicamento de referência e estabelecer seus atributos de qualidade críticos (CQAs), elaboramos um processo de produção para o biossimilar. Depois, após vários anos de testes, coletamos dados para provar a similaridade e obter aprovação.
- Uma comparação abrangente dos atributos da molécula biossimilar com o medicamento de referência usando tecnologia de ponta garante a ausência de diferenças clinicamente significativas.1,2
- Mesmo com dados analíticos robustos, o biossimilar somente pode ser aprovado com dados clínicos e não clínicos adicionais.2
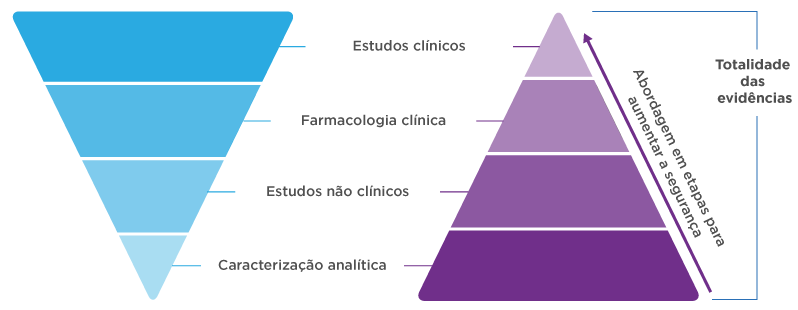
Desenvolvimento de biossimilares1,3
Demonstrar a segurança, a pureza e a potência em uma ou mais condições de uso apropriadas para as quais o medicamento de referência foi licenciado.
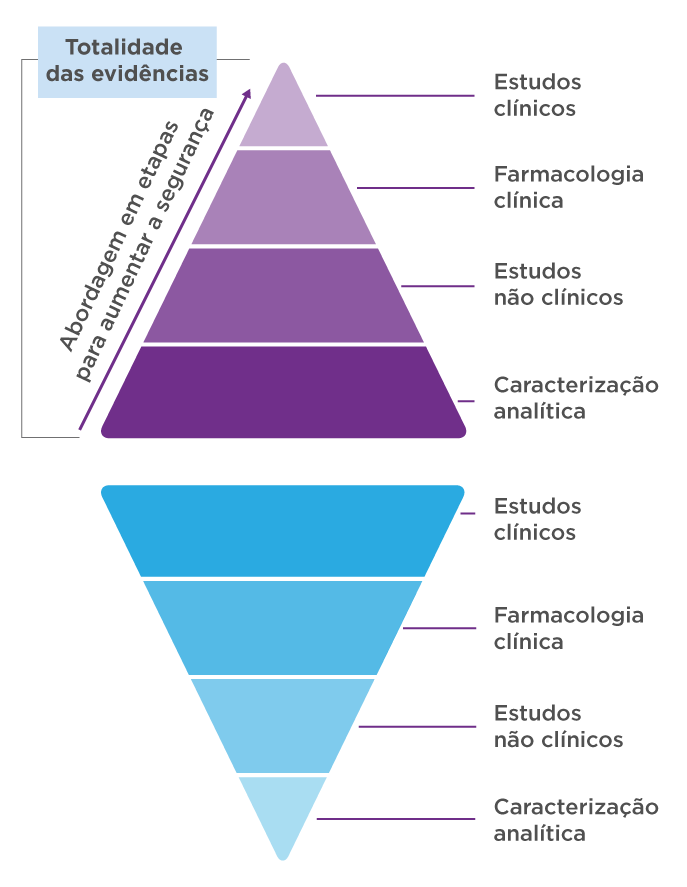
Desenvolvimento do medicamento de referência3
Demonstrar a segurança e a eficácia de um novo produto com evidências substanciais, adequadas e bem controladas.
A totalidade das evidências define a similaridade
Juntos, os dados acima compõem a totalidade das evidências usada para avaliar o nível de similaridade.1
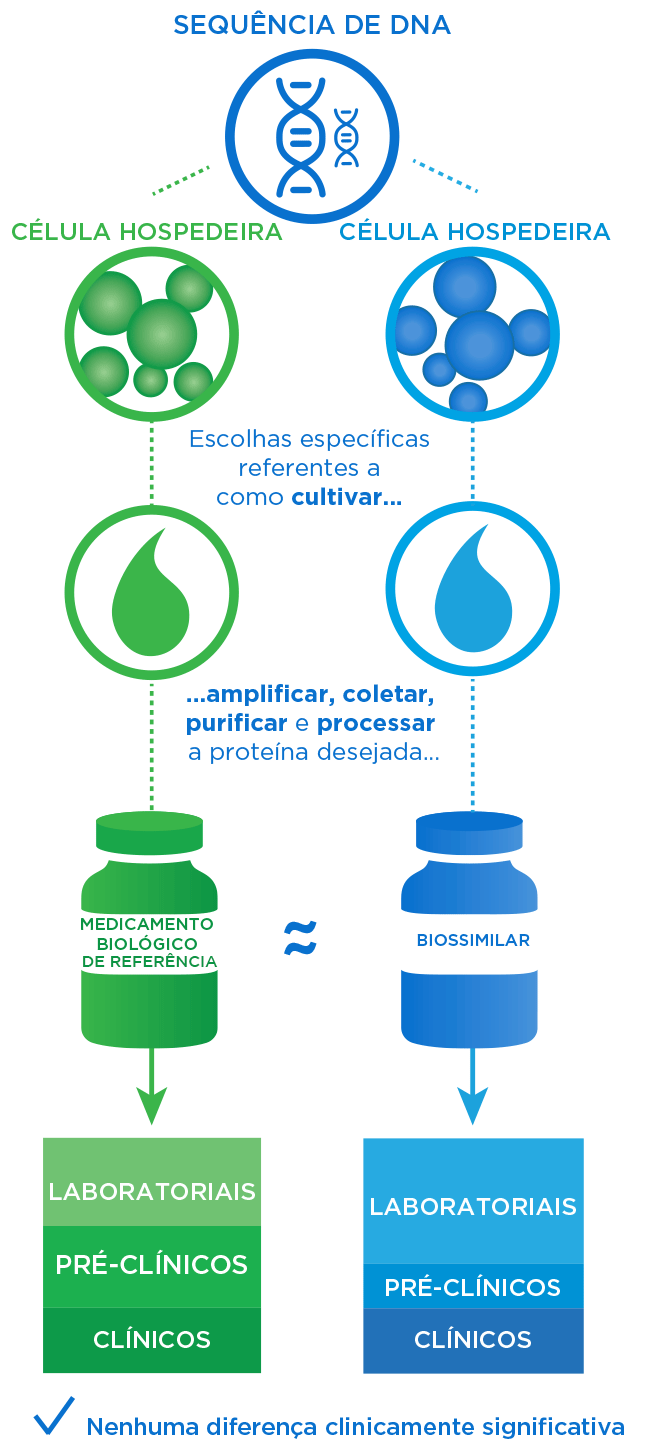
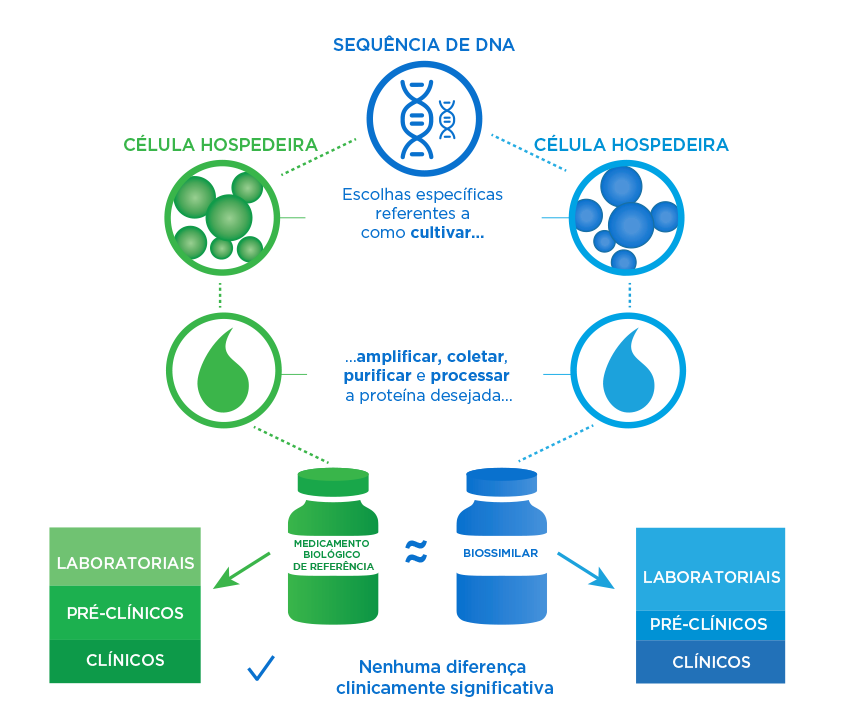
Juntos, os dados acima compõem a totalidade das evidências usada para avaliar o nível de similaridade.1

Dados clínicos
As exigências clínicas para o desenvolvimento de biossimilares diferem de acordo com a agência regulatória. De maneira geral, estudos de fase 1 e de fase 3 são obrigatórios.6,8 Estudos de fase 2 normalmente não são exigidos, uma vez que a dosagem do medicamento de referência já foi estabelecida.
Em estudos de fase 1, dados de farmacocinética (PK) e de farmacodinâmica (PD) são coletados de voluntários saudáveis ou pacientes. Eles são conduzidos de uma destas maneiras:6,7
- Grupos paralelos: participantes saudáveis recebem o biossimilar ou o medicamento biológico de referência
- Cruzados: dois grupos de participantes recebem primeiro o biossimilar ou o medicamento de referência; depois, os grupos são invertidos e passam a receber o outro tratamento
Estudos de fase 3 demonstram que o medicamento biossimilar tem eficácia, segurança e imunogenicidade similares às do medicamento de referência. Esses estudos devem ser realizados em uma população suficientemente sensível para possibilitar a detecção de diferenças clinicamente significativas entre o biossimilar e o medicamento de referência. Um estudo de fase 3 vai demonstrar:1
- Eficácia nem inferior nem superior à do medicamento de referência
- Segurança e imunogenicidade equivalentes
As exigências clínicas para o desenvolvimento de biossimilares diferem de acordo com a agência regulatória. De maneira geral, estudos de fase 1 e de fase 3 são obrigatórios.6,8 Estudos de fase 2 normalmente não são exigidos, uma vez que a dosagem do medicamento de referência já foi estabelecida.
Extrapolação
As agências reguladoras revisam a totalidade das evidências para determinar se o biossimilar pode ser aprovado para algumas ou para todas as indicações do produto de referência.1
References: 1. US Food and Drug Administration. Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. www.fda.gov/downloads/drugs/guidances/ucm291128.pdf. Published April 2015. Accessed December 21, 2018. 2. Conner J, Wuchterl D, Lopez M, et al. The biomanufacturing of biotechnology products. In: Shimasaki C, ed. Biotechnology Entrepreneurship: Starting, Managing, and Leading Biotech Companies. Waltham, MA: Academic Press; 2014:351-385. 3. Kozlowski S. US FDA perspectives on biosimilar biological products. Presented at: 2014 Biotechnology Technology Summit; June 13, 2014; Rockville, MD. www.ibbr.umd.edu/sites/default/files/public_page/Kozlowski%20-%20Biomanufacturing%20Summit.pdf. Accessed January 10, 2019. 4. Kresse GB. Biosimilars—science, status and strategic perspective. Eur J Pharm Biopharm. 2009;72:479-486. 5. European Medicines Agency. Guideline on Similar Biological Medicinal Products. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. Published October 23, 2014. Accessed January 10, 2019. 6. US Federal Trade Commission. Emerging Health Care Issues: Follow-On Biologic Drug Competition. www.ftc.gov/sites/default/files/documents/reports/emerging-health-care-issues-follow-biologic-drug-competition-federal-trade-commission-report/p083901biologicsreport.pdf. Published June 2009. Accessed December 21, 2018. 7. Alten R, Cronstein BN. Clinical trial development for biosimilars. Semin Arthritis Rheum. 2015;44:S2-S8. 8. Lai Z, La Noce A. Key design considerations on comparative clinical efficacy studies for biosimilars: adalimumab as an example. RMD Open. 2016;2:e000154.